Breaking Open the Black Box: Making Protein Structure Prediction Interpretable

Nithin Parsan
Co-Founder
Why Interpretability Matters
Large protein language models such as ESMFold and AlphaFold have reshaped our ability to predict three-dimensional structures directly from amino-acid sequences. Yet the nonlinear circuitry that powers these models remains opaque. Understanding how they work is essential for building trust, guiding design decisions, and uncovering new biological principles.
Our recent paper, "Towards Interpretable Protein Structure Prediction with Sparse Autoencoders," tackles this challenge head-on. Below, we highlight the core ideas, findings, and resources from the work in an accessible recap.
Two Key Advances
- Scaling sparse autoencoders to ESM2-3B. By training sparse autoencoders (SAEs) on internal representations of ESM2-3B—the base model behind ESMFold—we open the door to mechanistic interpretability for state-of-the-art structure prediction.
- Introducing Matryoshka SAEs for hierarchical features. Our architecture forces nested groups of latents to reconstruct inputs independently, yielding multi-scale features that mirror protein biology.
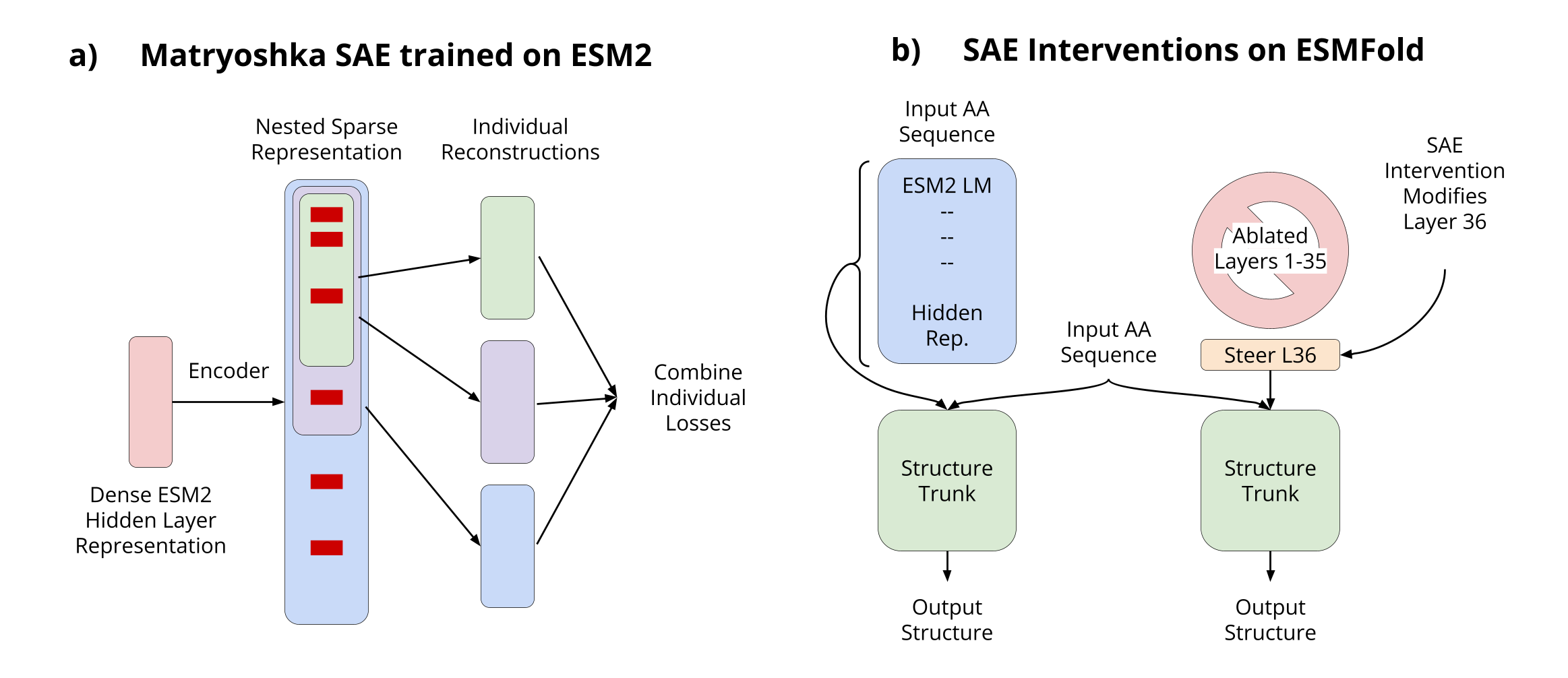
Figure 1. Schematic of our Matryoshka SAE approach.
Methods in Brief
We train SAEs on hidden activations from every layer of ESM2-3B. To test whether these compressed, sparse representations preserve function, we evaluate them on:
- Language modelling loss (ΔCE)
- Structure prediction with ESMFold via targeted layer ablation
- Biological concept discovery across 476 Swiss-Prot annotations
- Unsupervised contact-map prediction using categorical Jacobians
What We Learned
- Scale is critical. Moving from 8 M to 3 B parameters nearly triples the fraction of biological concepts captured by single features (≈49 % coverage).
- Only a few latents are needed. With as few as 8-32 active features per token, SAEs recover structure-prediction performance, hinting at compact underlying rules.
- Hierarchical organization helps. Matryoshka SAEs match or exceed standard SAEs while providing clear multi-scale groupings.
- Features are causal. By steering solvent-accessibility-related features, we can systematically tune ESMFold's structural outputs without changing the input sequence.
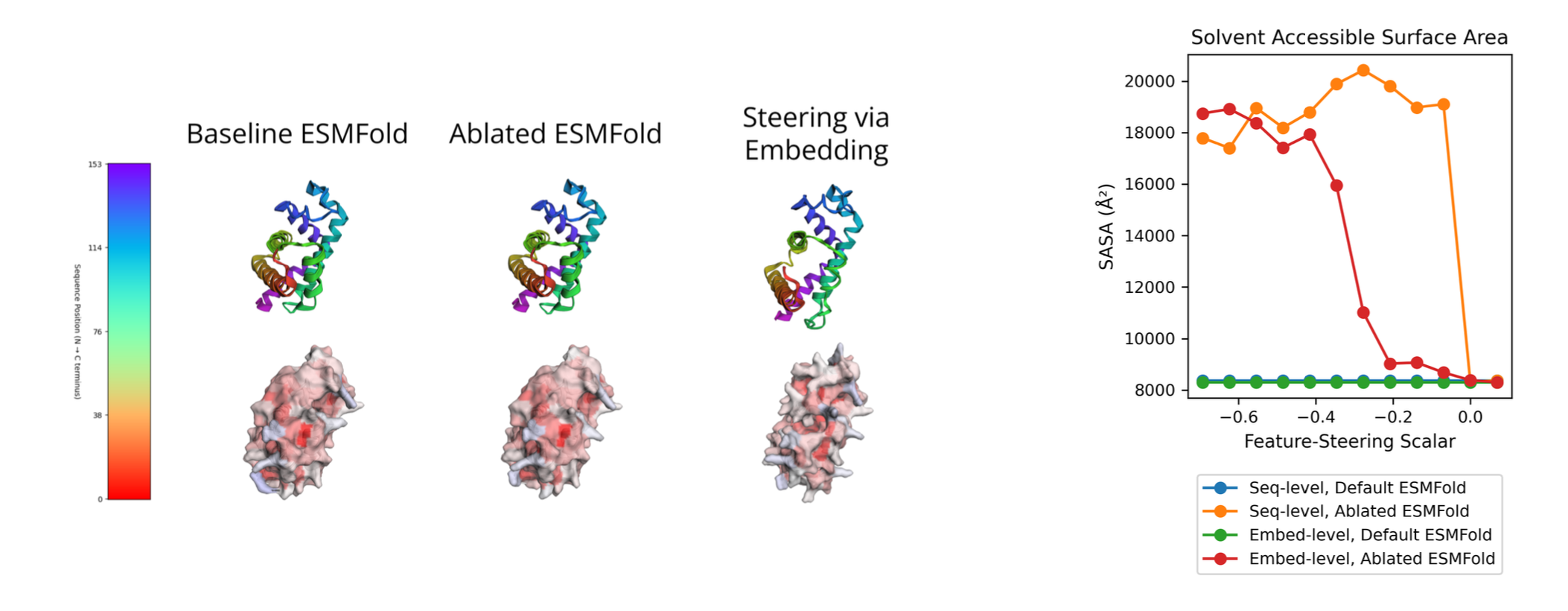
Figure 2. Steering a solvent-accessibility feature changes predicted surface exposure.
Explore the Interactive Visualizer
We built a public web tool that lets you browse the hierarchical features learned by our SAEs, highlight them on sequences, and see corresponding 3-D structures. Give it a spin and explore how specific patterns map to structural motifs.
Looking Ahead
Interpretability is more than an academic exercise—it provides the levers we need to control and trust large biological models. By revealing how sequence information flows to structure, we hope to accelerate principled protein design across many applications.
All code, trained models, and detailed benchmarks are openly available on GitHub.
Schedule a call with us today to learn more.
Book a CallCitation: Parsan, N., Yang, D.J., & Yang, J.J. (2025). Towards Interpretable Protein Structure Prediction with Sparse Autoencoders. ICLR GEM Bio Workshop 2025.