Polygenic Prediction: The Problem, the Landscape, and What We're Building

John Yang
Co-Founder & CEO

Complex traits are 50–90% heritable according to twin and family studies, but the best deployed polygenic models explain only a fraction of that variance. The gap is large, well-documented, and — we believe — partly a problem of model class. Current polygenic risk scores are linear models trained on GWAS summary statistics. They cannot represent haplotype context, epistatic interactions, or regulatory grammar.
At Reticular, we're working on models that narrow this gap, starting with embryo selection — a setting where prediction accuracy has direct, irreversible consequences.
This post covers the problem, the current landscape, and what we're building.
Context: embryo selection and the market
IVF is a $25B global industry ($3.6B in the US), with ~400K cycles/year in the US and growing. The market has expanded steadily as average maternal age rises, employer benefits packages increasingly cover fertility treatments, and social egg freezing normalizes the technology for a broader demographic. It is primarily a self-pay, cash market — a single cycle typically costs $15–35K out of pocket, and most insurance plans do not cover it. Over 60% of IVF patients have household incomes above $100K. This means the customer base is already accustomed to paying significant sums for reproductive outcomes, without insurance intermediation.
~55% of cycles already include some form of preimplantation genetic testing (PGT), so there is an established and receptive customer base paying for genetic information about their embryos. Polygenic embryo screening (PGT-P) is the next layer: scoring embryos across complex heritable diseases and traits. The decision to implant one embryo over another is irreversible, so prediction accuracy matters more here than in most genomics applications.
The deeper opportunity is market expansion. Today, IVF is primarily pursued by couples with fertility challenges. But if polygenic prediction becomes accurate enough to meaningfully reduce disease risk in offspring, a large new population has strong motivation to pursue IVF electively: people with significant family histories of heritable conditions — schizophrenia, bipolar disorder, early-onset Alzheimer's, hereditary cancers, severe cardiovascular risk. For this population, embryo selection is not a fertility intervention; it is a health decision. Solving the prediction problem doesn't just improve an existing product — it creates a new category of patients who would otherwise never have considered IVF.
Three primary competitors — Nucleus ($32M raised), Orchid ($16.5M), and Herasight — have raised $50M+ combined. No one has established clear technical superiority yet, and all are using variants of the same underlying model class (see below).
Competitor pricing
| Provider | Price | Structure | Method |
|---|---|---|---|
| Herasight | ~$50K | Per analysis | SBayesRC, within-family validation focus |
| Nucleus | ~$10K | Per embryo set | SBayesRC, broadest disease coverage |
| Orchid | ~$2,500/embryo (~$25–50K/cycle) | Per embryo | Older PRS pipelines, 30× WGS |
| Genomic Prediction | ~$2,000/embryo | Per embryo | PRS |
The 20× spread in list price across providers using essentially the same underlying model class is itself a signal: no one has established a clear accuracy advantage, so there is no settled pricing anchor. The market is waiting for a technical differentiation story.
The venture case
The math is straightforward. ~400K IVF cycles happen in the US each year. At 20% PGT-P attach and a $10K average selling price, that is an ~$800M US revenue opportunity. Globally, with 2M+ annual IVF cycles, the addressable market is multi-billion.
Three structural features make this unusually attractive:
- Cash pay, no insurance. One of the only large healthcare markets that is almost entirely self-pay. No reimbursement negotiation, no 18-month payer sales cycle. The customer is the patient, and they are already writing large checks — a single IVF cycle costs $15–35K out of pocket. Adding $10–50K for genetic screening is a natural upsell into an existing high-WTP relationship.
- No FDA approval required. Polygenic screening falls under laboratory-developed tests (LDTs). Companies are already selling this product commercially today without a regulatory pathway. Speed to market is determined by science and clinical partnerships, not agency timelines.
- Winner-take-most clinic dynamics. IVF clinics will converge on one or two validated providers. Herasight already charges $50K vs Orchid's ~$2,500/embryo — same underlying model class, different positioning. The first to demonstrate rigorous within-family validated accuracy improvement can command premium pricing across the entire market and lock in clinic relationships before competitors catch up.
How the product works
Because IVF is already a direct-to-consumer cash market, polygenic screening slots naturally into the existing payment flow. The product is sold directly to parents through IVF clinics, at price points currently ranging from ~$2.5K per embryo to ~$50K per analysis depending on the provider.
The workflow is straightforward: sequence the parents and the embryos (from biopsied cells already taken during standard PGT), run polygenic scores across conditions, and return risk reports to the patient. Clinicians sign off on the results, and genetic counselors walk parents through the scores and their implications before any implantation decision is made.
This is not drug discovery. There is no FDA approval process for polygenic scoring — it falls under laboratory-developed tests (LDTs). Companies are already selling this product commercially today.
Why parents buy imperfect models
Parents going through IVF are making an irreversible decision on a fixed timeline — they are having a child regardless, and typically need to choose which embryo to implant within weeks. They are not waiting for a perfect predictor. They want the most informed decision possible given the available science. Even a model that explains a modest fraction of heritable variance provides information that would otherwise be unavailable, and parents are willing to pay for that edge when the stakes are this high. The bar is not "perfect prediction" — it's "better than choosing blind."
How do we know better prediction is possible?
Twin and family studies show substantial heritable signal for complex traits,[6] but today's best polygenic scores capture only a fraction — the classic missing heritability problem.[1]
| Trait | Twin h² (broad) | SNP h² | Best PGS R² | Missing Gap (Twin h² − PGS R²) |
|---|---|---|---|---|
| Height | ~0.80 | ~0.45–0.50 | ~0.40 (SBayesRC, 7M SNPs, n≈700K) | ~0.40 |
| Schizophrenia | ~0.73–0.81 | ~0.20–0.25 | ~0.07–0.10* | ~0.63–0.74 |
| Major Depression (MDD) | ~0.37–0.40 | ~0.09–0.13 | ~0.02–0.04 | ~0.33–0.38 |
| Bipolar Disorder | ~0.60–0.85 | ~0.20–0.25 | ~0.04–0.08 | ~0.52–0.81 |
| Type 2 Diabetes | ~0.50–0.70 | ~0.18–0.20 | ~0.06–0.10 | ~0.40–0.64 |
| Coronary Artery Disease | ~0.40–0.60 | ~0.10–0.15 | ~0.04–0.08 | ~0.32–0.56 |
| ADHD | ~0.70–0.80 | ~0.14–0.22 | ~0.03–0.06 | ~0.64–0.77 |
| Autism (ASD) | ~0.64–0.91 | ~0.12–0.20 | ~0.02–0.06 | ~0.58–0.89 |
| Intelligence / GCA | ~0.50–0.80 | ~0.20–0.25 | ~0.10–0.16 | ~0.40–0.64 |
| BMI | ~0.70 | ~0.25–0.30 | ~0.16 (SBayesRC, 7M SNPs) | ~0.54 |
Continuous traits (Height, Intelligence, BMI) use observed-scale R². *Binary trait PGS values are reported on the liability scale (liability R²), which transforms observed R² to a hypothetical continuous liability scale using R²l = R²obs × K²(1−K)² / [z²P(1−P)], where K is population prevalence, z is the normal density at the liability threshold, and P is sample prevalence. This places PGS accuracy and heritability on the same scale so the gap column is directly comparable across all traits.
The gap between twin heritability and current PGS accuracy is substantial across nearly every trait. Some of this gap may reflect non-additive or environmental factors that genotype-only models cannot recover[7] — but the size of the gap, and the fact that SNP h² itself exceeds current PGS accuracy, suggests meaningful room for methodological improvement.
What "state of the art" looks like today
The industry baseline is SBayesRC — a Bayesian model that builds polygenic predictors from GWAS summary statistics with functional annotations across ~7M common SNPs.[2]
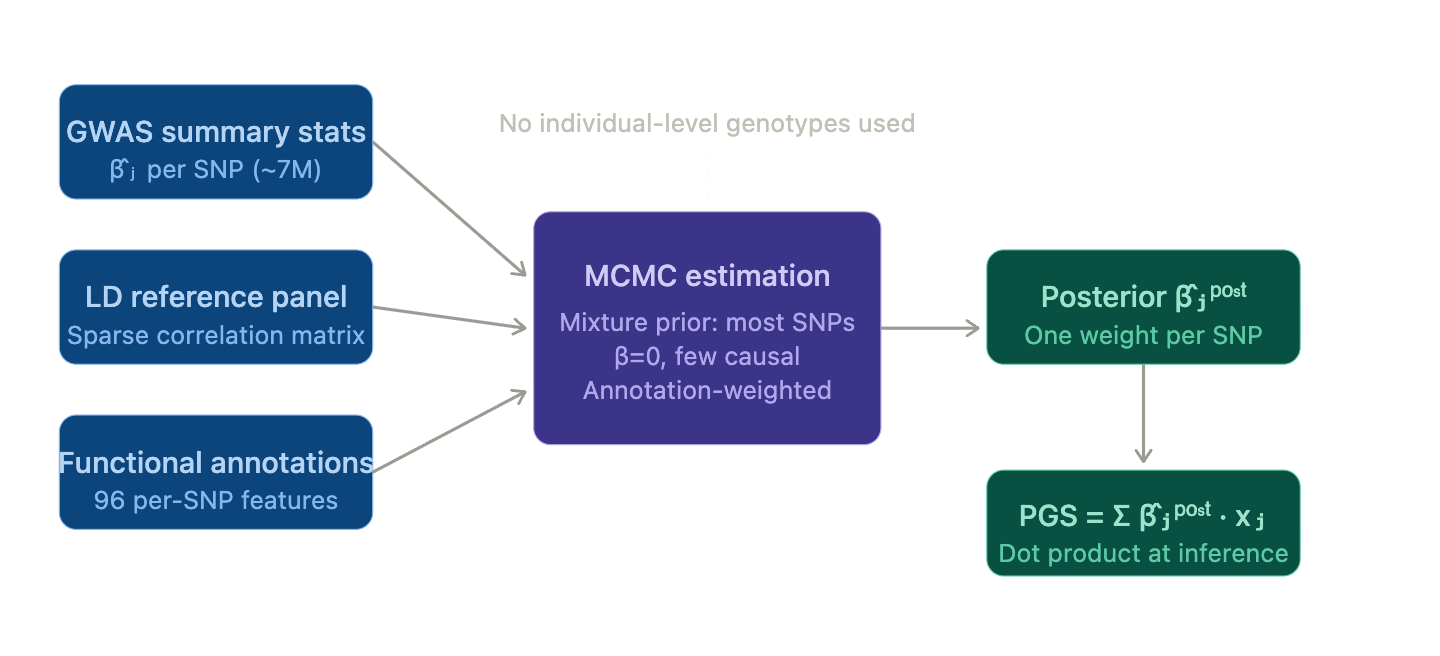
SBayesRC model architecture and annotation framework.
SBayesRC fits a Bayesian mixture prior (most SNPs zero, small fraction causal), with an annotation layer modulating each SNP's prior probability of being causal. Training runs MCMC; inference is a dot product. It represents meaningful improvement over earlier methods, and is what powers the commercial landscape today:
- Herasight — SBayesRC-based, focused on within-family validation. Charges $50K.
- Nucleus — SBayesRC-based, broadest disease coverage. Charges $10K.
- Orchid — older PRS pipelines, 30× whole-genome sequencing. Charges $2,500 per embryo ($25K–$50K).
| Disease | Herasight | Nucleus | Orchid | Genomic Prediction |
|---|---|---|---|---|
| Type 2 Diabetes | 20.7% (AUC ~0.724) | 22.9% (AUC ~0.736) | 5.0% (AUC ~0.610) | 5.5% (AUC ~0.615) |
| Prostate Cancer | 19.0% (AUC ~0.730) | 21.6% (AUC ~0.745) | 12.3% (AUC ~0.686) | 6.7% (AUC ~0.637) |
| Breast Cancer | 14.3% (AUC ~0.697) | 12.8% (AUC ~0.687) | 8.8% (AUC ~0.655) | 3.3% (AUC ~0.595) |
| Hypertension | 15.7% (AUC ~0.681) | 15.5% (AUC ~0.680) | — | — |
| Alzheimer's Disease | 16.1% (AUC ~0.716) | 21.1% (AUC ~0.747) | — | — |
| Coronary Artery Disease | — | 21.0% (AUC ~0.712) | — | — |
Company-reported liability R² values. Parenthetical AUCs are implied estimates derived from each liability R² using the Wray et al. liability-threshold approximation[8] with disease-specific lifetime prevalence. Liability R² is prevalence-dependent — the same discrimination ability yields a lower R² for rarer diseases — so the implied AUC makes cross-disease and cross-provider comparisons more interpretable. These AUCs are approximations and should not be treated as directly validated.
SBayesRC represents the current ceiling of summary-stat PRS methods. Its structural constraints are worth noting explicitly:
- Summary-stat training only
- Mostly additive / linear effect structures
- Common-variant focus
- No ability to exploit haplotypes, multi-allelic variation, regulatory context, or non-additivity
Technical gaps and our hypotheses
1) Additivity + weak tail behavior
PRS methods are fundamentally additive-effect models. They tend to shrink extreme predictions toward the mean. In embryo selection, the decision-relevant variation is often in the tails (high-risk embryos, rare disease liability), so tail calibration matters more than average-case R².
PRS models shrink extreme predictions toward the mean, reducing tail fidelity where embryo selection decisions concentrate.
Direction we're exploring: Objectives and architectures that preserve tail fidelity — calibrated uncertainty, tail-aware losses, distributional prediction. Whether this yields meaningful improvement over well-calibrated linear baselines is an empirical question we're actively testing.
2) Summary-stat training vs. individual-level learning
SBayesRC trains entirely on summary statistics because they're easy to share. But this constrains what you can learn: no rich covariates, no interaction structure, no family structure, limited flexibility in the learning objective.
Why this is changing now: UK Biobank's ~500K whole-genome sequences make population-scale, individual-level training practical for the first time, including phased haplotypes, rare variants, and structural variation, alongside deep phenotyping. UKB is also releasing matched molecular readouts, like Olink blood proteomics (~3K proteins across ~50K participants), with tissue-specific RNA-seq expected. Together, this enables training approaches that summary statistics cannot support.
Direction we're exploring: With this individual-level data, use modern ML training loops — pairwise ranking losses, representation learning, richer targets — while retaining the causal and robustness lessons from statistical genetics. The risk is overfitting; the opportunity is learning signal that summary stats structurally discard.
3) Input representation misses real genomic structure
The standard featurization ("millions of independent-ish SNPs as dosages") is a convenience abstraction. It can't naturally capture haplotype context, epistatic effects, richer variant classes, or regulatory grammar.
Direction we're exploring: Learn representations over phased haplotype blocks and incorporate functional priors, while keeping the approach scalable. It's an open question how much of the heritability gap is attributable to non-additive or haplotype-level effects versus simply needing larger additive models with better regularization.
4) Sequence-to-function models as inductive bias
A complementary path is to model intermediate biology — predict molecular/functional consequences from sequence, then feed those into phenotype prediction. This can improve generalization (shared biology transfers across cohorts), handle rare variants more naturally, and provide interpretability.
Recent relevant work: CZI's VariantFormer (1.2B-parameter hierarchical transformer, trained on 21K genome–transcriptome pairs) achieves SOTA on personalized expression prediction by explicitly encoding individual variant profiles with tissue-specific conditioning — outperforming both population-based methods and large DNA foundation models (Evo2) without explicit variant encoding.[3]
Important caveat: Even strong sequence-to-function models can underperform simple genotype baselines for personal transcriptome prediction on some modalities.[4] We think of these models as variant annotation priors and representation learners rather than drop-in replacements for genotype-based predictors. Whether they add incremental R² on top of strong PRS baselines for trait prediction is not yet established.
5) Evaluation: population-level accuracy is misleading
There's a known gap between population-level associations and within-family predictive utility (stratification, assortative mating, indirect genetic effects). Highly parameterized PGS can overfit to population structure and pass standard evaluations while failing null hypothesis tests.[5]
Our position: Within-family validation should be a core metric for any claim meant to support embryo selection. We use it as our primary evaluation standard.
Research directions
Near-term (0–6 months)
- Individual-level training with modern ML objectives — replace summary-stat pipelines with direct training on UKB individual-level data: pairwise ranking losses, tail-aware objectives, calibrated uncertainty heads
- Haplotype-aware input representations — phased haplotype encodings and learned embeddings over local haplotype blocks
- Rare variant integration — incorporate rare coding variants (pLOF, missense) alongside common SNPs via WGS-derived burden scores
- Sequence-to-function priors — annotate variants with Borzoi/AlphaGenome regulatory impact scores as auxiliary features, benchmark incremental lift
- Publishable within-family validation study — the credibility anchor for commercial deployment, targeting a top-tier genetics journal
Longer-term (6–24 months)
- Multi-ancestry generalization — train and evaluate on non-European biobank cohorts with transfer and calibration methods
- Variant-to-phenotype pretraining — pretrain a transformer on phased VCFs to learn richer per-variant representations, fine-tune on phenotype prediction; corpus extends beyond SNPs to indels, SVs, and functional annotations
- Multimodal integration — incorporate EHRs, blood proteomics, tissue-specific RNA-seq, and imaging from UKB as training signal
Why we think this is worth working on
Over $50M has been deployed into polygenic embryo screening without a clear technical differentiation — the leading companies are all using variants of the same linear PRS methodology. The question of whether modern deep learning can meaningfully improve polygenic prediction under rigorous evaluation (especially within-family) is genuinely open. We think it can, but we hold that as a hypothesis to be tested, not a foregone conclusion.
If this problem interests you — sequence-to-phenotype prediction at the intersection of deep learning and human genomics, with direct clinical consequences — we'd like to talk.
If this problem interests you — sequence-to-phenotype prediction at the intersection of deep learning and human genomics, with direct clinical consequences — reach out at john@reticular.com.
References
- Visscher. Solving the missing heritability problem (PLOS Genetics, 2019). [link]
- Zeng et al. Leveraging functional genomic annotations and genome coverage to improve polygenic prediction (SBayesRC; Nature Genetics, 2024). [link]
- Ghosal et al. VariantFormer: A hierarchical transformer integrating DNA sequences with genetic variations and regulatory landscapes for personalized gene expression prediction (bioRxiv, 2025). [link]
- Tu et al. Modality gap in personal-genome prediction (bioRxiv, 2026). [link]
- Aw et al. Overfitting and confounding in modern PGS (PMC, 2024). [link]
- Polderman et al. Meta-analysis of twin studies (Nature Genetics, 2015). [link]
- Turkheimer & Matthews. Three Legs of the Missing Heritability Problem (2022). [link]
- Wray et al. The genetic interpretation of area under the ROC curve in genomic profiling (PLoS Genetics, 2010). [link]